|
|
| שורה 8: |
שורה 8: |
| | |ICD-9={{ICD9|272.7}} | | |ICD-9={{ICD9|272.7}} |
| | |MeSH={{MeSH|D000795}} | | |MeSH={{MeSH|D000795}} |
| − | |יוצר הערך=ד"ר אניק רז-רוטשילד | + | |יוצר הערך=פרופסור אניק רז-רוטשילד |
| | |אחראי הערך= | | |אחראי הערך= |
| | }} | | }} |
| | {{הרחבה|מחלות אגירה}} | | {{הרחבה|מחלות אגירה}} |
| − | '''מחלת אנדרסון-פברי''' היא מחלת אגירה ליזוזומלית המורשת בתאחיזה לכרומוזום ה-X{{כ}} (X- linked). תפוצתה באוכלוסיה הנה פאן-אתנית. המאפיין הביוכימי של מחלת פברי הנו ירידה או חסר בפעילות של האנזים אלפא גלקטוזידאז A {{כ}} (Alpha-galactosidase-A) הגורם לאגירה של סובסטרט ממשפחת הגליקוספינגוליפידים (Glycosphingolipids) ופגיעה במערכות שונות של הגוף. | + | '''מחלת אנדרסון-פברי''' (Anderson-Fabry) היא מחלת אגירה ליזוזומלית המורשת בתאחיזה לכרומוזום ה-X {{כ}}(X- linked). תפוצתה באוכלוסיה היא פן- אתנית ושכיחותה אינה ברורה. |
| | | | |
| − | הפגיעה העיקרית כוללת את מערכת העצבים הפריפרית והמרכזית, מערכת הלב וכלי הדם (קרדיווסקולרית) והכליות. משמעותה של הורשה אחוזה ל-X הינה כי הגברים הנם המיזיגוטים (Hemizygotes) (בעלי כרומוזום X בלבד הנושא את המוטציה) ומפתחים מחלה רב מערכתית, ואילו הנשים הן הטרוזיגוטיות (בעלות כרומוזום X הנושא את המוטציה, וכרומוזום X שאינו נושא את המוטציה) אך על פי רוב תפתחנה גם הן תסמינים עם השנים. זוהי מחלה פרוגרסיבית וסימניה הראשונים מופעים כבר בגיל הילדות (1,2). חומרת המחלה וצורת הופעתה מגוונות בין המשפחות השונות וגם בין חולים של אותה משפחה. משנת 2001 זמין הטיפול באנזים חליפי ועל כן, אבחון מוקדם ומתן טיפול בטרם נוצר נזק בלתי הפיך לרקמה ולאברים עשויים לשפר את איכות חיי החולים ואולי אף להאריכם (3,4).
| + | מחלת פברי מאופיינת על ידי ירידה או חסר בפעילות של האנזים הליזוזומלי alpha-galactosidase-A המובילה לאגירה של סובסטרט ממשפחת הגליקוספינגוליפידים ולפגיעה רב מערכית. מחלת Fabry היא חלק מקבוצת הסיפינוליפידוזות .(sphingolipidosis) הפגיעות העיקריות הן במערכת העצבים, במערכת הקרדיווסקולרית ובכליות. הגברים הינם המיזיגוטים (hemizygotes) ומפתחים מחלה רב מערכתית ואילו הנשים הן הטרוזיגוטיות אך על פי רוב תפתחנה גם הן תסמינים אשר עלולים להיות חמורים כמו בגברים. |
| | | | |
| − | ==אפידמיולוגיה==
| + | זוהי מחלה פרוגרסיבית וסימניה הראשונים יכולים להופיע כבר בגיל הילדות. חומרת המחלה וצורת הופעתה מגוונות בין המשפחות השונות וגם בתוך כל משפחה. |
| | | | |
| − | מחלת אנדרסון-פברי אינה שכיחה יותר בעדות או עמים מסוימים ופוגעת בכל האוכלוסייה. בעבר תוארה המחלה כמחלה נדירה המופיעה בזכרים בשכיחות של 1:40,000-60,000 (1). אולם, במחקר סקר ילודים שנערך באיטליה נמצאה פעילות ירודה של האנזים אלפא גלקטוזידאז A (מתחת ל-20% מהנורמה) בשכיחות של 1:3100 בתינוקות ממין זכר (13) ובסקר שנערך בטאיוואן נמצאה שכיחות של 1:1250, כאשר 86% מהחולים נשאו אותה מוטציה אופיינית אשר קשורה להופעה מאוחרת של המחלה (14). בישראל מאובחנים כיום כ-25-30 חולים (גברים, נשים וילדים).
| + | זה כבר יותר מ-14 שנה קיים טיפול באנזים חליפי ועל כן, אבחון מוקדם ומתן טיפול, בטרם נוצר נזק בלתי הפיך לרקמה ולאיברים, עשויים לשפר את איכות חיי החולים ואולי אף להאריכם {{הערה|שם=הערה1|}}. |
| | | | |
| − | ==אטיולוגיה==
| + | שמות נוספים: Fabry disease ,Fabry’s disease ,Alpha-galactosidase A deficiency, Angiokeratoma corporis diffusum, Ceramide trihexosidosis, Ruiter-Pompen-Wyers syndrome, Sweeley-Klionsky disease. |
| | | | |
| − | ===פתופיזיולוגיה===
| |
| | | | |
| − | בעקבות חסר או ירידה בפעילות האנזים אלפא גלקטוזידאז A נמנע פירוק תקין של גליקוספינגוליפידים (Glycosphingolipids) ובעיקר של Globotriaosylceramide{{כ}} (GL3,Gb3, ceramide trihexoside) בתאים שונים ובמיוחד בתאי האנדותל הוסקולרי. הסובסטרט נאגר בליזוזומים וגורם לפגיעה בתפקוד תאים כגון תאי עצב, תאי האפיתל הגלומרולרי (Glomerular epithelia) והטובולרי (Tubular epithelia) של הכליות ותאי שריר הלב (קרדיומיוציטים) (1). הפגיעה התאית גוררת פגיעה ברקמה עם הופעה של [[איסכמיה]] (Ischemia) ו[[פיברוזיס]] (Fibrosis). כיום עדיין לא ברור באיזה שלב הנזק לתאים הוא בלתי הפיך ומתי הטיפול אינו יכול להביא לשיפור במצבם של החולים. לאחרונה מצטברות עדויות כי מתן טיפול מוקדם ככל האפשר הנו יעיל יותר ואף עשוי למנוע סיבוכים של המחלה כגון [[כשל כלייתי]], [[שבץ מוחי]] או [[התקף לב]] בגיל צעיר (3,4,5).
| + | ==פתופיזיולוגיה== |
| | | | |
| − | ===תורשה=== | + | על פי הידוע נכון להיום, בעקבות חסר או ירידה בפעילות האנזים alpha-galactosidase-A נמנע פירוק תקין של גליקוספינגוליפידים (Glycosphingolipids) ובעיקר Globotriaosylceramide{{כ}} (GL3,Gb3, ceramide trihexoside) בתאים שונים ובמיוחד בולט הדבר בתאי האנדותל הווסקולרי. הסובסטרט נאגר בליזוזומים וגורם לפגיעות משניות בתפקוד תאים כגון תאי עצב, תאי האפיתל הגלומרולרי (glomerular epithelia) והטובולרי (tubular epithelia) של הכליות, תאי מערכת ההולכה של קוצבי הלב וקרדיומיוציטים (cardiomyocytes)נ(1). הפגיעה התאית גוררת פגיעה ברקמה עם הופעה של איסכמיה (ischemia) ופיברוזיס (fibrosis). עד היום לא ברור מתי הנזק לתאים הוא בלתי הפיך ומתי הטיפול אינו יכול להביא לשיפור במצבם של החולים. האגירה מתחילה כבר בשלב העוברי {{הערה|שם=הערה2|}} ומצטברות עדויות כי מתן טיפול מוקדם הוא יעיל {{הערה|שם=הערה1}}, {{הערה|שם=הערה5|}, {{הערה|שם=הערה6|}}. |
| | | | |
| − | הגן המקודד לאנזים אלפא גלקטוזידאז A ממופה על גבי הזרוע הארוכה של כרומוזומום X{{כ}} (Xq22) ומכיל 7 אקסונים שהם האזורים המקודדים לאנזים (6). לכל משפחה מוטציה משלה (Private mutation). עד היום תוארו יותר מ-384 מוטציות (7). מחלת אנדרסון-פברי מורשת בתאחיזה לכרומוזום ה-X. חשוב לציין כי נשים הנושאות מוטציות בגן לאלפא גלקטוזידאז A יהפכו להיות עם הזמן סימפטומטיות בדרגות חומרה שונות (8,9). הסיבה להופעת המחלה בנשים לא ברורה אך דפוס של הופעת סימפטומים בנשים במחלה שאינה דומיננטית ומופיעה בתאחיזה לכרומוזום ה-X אינו ייחודי למחלת אנדרסון-פברי (10,11, 12). לאישה נשאית יש סיכון של 50% לילד זכר חולה, ו-50% לילדה נשאית אשר יכולה לפתח תסמנים בהמשך. בנותיו של גבר חולה אנדרסון-פברי בהכרח נשאיות ואילו כל בניו יהיו בריאים.
| + | ==תורשה ושכיחות== |
| | | | |
| − | [[קובץ:פבריתרשים1.JPG|מרכז|ממוזער|400px|תרשים מספר 1]] | + | הגן המקודד לאנזים alpha-galactosidase-A ממופה על גבי הזרוע הארוכה של כרומוזום X {{כ}}[Xq22] ומכיל 7 אקסונים {{הערה|שם=הערה5}}. לכל משפחה מוטציה משלה (private mutation). מחלת פברי מורשת בתאחיזה לכרומוזום ה-X. בניגוד למחלות תורשתיות אחרות המועברות בתאחיזה לכרומוזום ה-X, שבהן הנשים הנשאיות בדרך כלל אינן סימפטומטיות, נשים הנושאות מוטציות בגן ל-Alpha galactosidase-A יהיו עם הזמן סימפטומטיות בדרגות חומרה שונות {{הערה|שם=הערה1}}. כמו כן, בשל דפוס ההורשה האחוז ל-X, תהיינה בנותיו של גבר חולה פברי בהכרח נשאיות ואילו כל בניו יהיו בריאים. לגבי אישה נשאית, לכל בת יהיה סיכוי של 50% להיות נשאית עם אפשרות לפתח את המחלה, כאשר לכל בן של אישה נשאית, הסיכוי לבן חולה עומד על 50%. |
| | | | |
| − | [[קובץ:פבריתרשים2.JPG|מרכז|ממוזער|400px|תרשים מספר 2]]
| + | מחלת Fabry אינה שכיחה יותר בעדות או בעמים מסוימים ופוגעת בכל האוכלוסיה בשכיחות דומה. בעבר תוארה המחלה כמחלה נדירה המופיעה בזכרים בשכיחות של 1:40,000-60,000 {{כ}}{{הערה|שם=הערה1}}, אולם במחקרי סקר ילודים שנערכו במספר מדינות, נמצאה שכיחות גבוהה בהרבה 1:1237-1:13,341 {{כ}}{{הערה|שם=הערה8|}}. בישראל מאובחנים כיום כ-30-25 חולים (גברים, נשים וילדים). |
| | | | |
| − | ==קליניקה==
| + | סימני המחלה |
| | + | רוב התסמינים שמהם סובלים חולי פברי אינם ספציפיים למחלה ולכן אבחון חולה עם פברי ואבחון חולים עם מחלות נדירות בכלל מהווים אתגר משמעותי. |
| | | | |
| − | חולה אנדרסון-פברי יופיע במרפאה עם אחד או יותר מהממצאים הבאים: נוכחות נגעים עוריים מסוג אנגיוקרטומה (Angiokeratoma), כאבים "שורפים" בגפיים (Acroparesthesia), כאבי בטן, שלשולים במיוחד אחרי אכילה, חוסר הזעה, עייפות, אי סבילות לחום, אי סבילות למאמץ וכן סיפור משפחתי של [[אי-ספיקת כליות]], [[שבץ מוחי]] (Stroke), או מחלה לבבית (1,2). אחד הכלים היעילים ביותר לאבחון המחלה הנו עריכת בירור אם קיימים עוד קרובי משפחה עם תסמינים אשר יכולים להיות קשורים למחלת אנדרסון-פברי. כלומר, בניית עץ משפחה המתאים לתורשה בתאחיזה ל-X. לאחר אבחון של אדם במשפחה חדשה יש צורך לבדוק את כל בני המשפחה המצויים בסיכון על בסיס נתוני המשפחה תוך התייחסות לאופן ההורשה. | + | חולה פברי יופיע במרפאה עם אחד או יותר מהממצאים הבאים (תמונה מס' 1): נוכחות נגעים עוריים מסוג angiokeratoma, כאבים "שורפים" בגפיים (acroparesthesia), כאבי בטן, שלשולים במיוחד אחרי אכילה, חוסר הזעה, עייפות, אי סבילות לחום, אי סבילות למאמץ וכן סיפור משפחתי של תסמינים כמו אי ספיקת כליות, שבץ מוחי (stroke), או מחלה קרדיאלית (1). |
| | | | |
| − | סימני המחלה מתחילים להופיע בגיל הילדות ומחמירים עם הזמן גם בגברים וגם בנשים (15). סימנים ראשונים מופיעים בבנים כבר בגילאים 3-10 ובבנות סביב גיל 13-19. האבחנה הסופית הנה בממוצע סביב גיל 26 בגברים וסביב גיל 30-32 בנשים (8, 9, 16). בתקופה זו ולמעשה עוד בשלבים העובריים מצטבר GL3 ברקמות ונוצר נזק שעלול להיות בלתי הפיך בגיל מבוגר יותר. חולים עם פעילות אנזימתית ירודה או לא קיימת, מציגים תמונה קלאסית של המחלה ואילו חולים עם פעילות אנזימתית שאריתית מציגים מחלה קלה יותר או Variant disease{{כ}} (17,19).
| + | אחד הכלים היעילים ביותר לאבחון המחלה הוא בירור האם קיימים קרובי משפחה עם תסמינים אשר יכולים להיות קשורים למחלת פברי. כלומר, בניית עץ משפחה המתאים לתורשה בתאחיזה ל-X שבו נמצא חולים עם התסמינים המוזכרים שהקשר ביניהם הוא הנשים במשפחה. לאחר אבחון של אדם במשפחה חדשה, יש צורך לבדוק את כל בני המשפחה המצויים בסיכון על בסיס נתוני המשפחה, תוך התייחסות לאופן ההורשה. לשם כך מומלץ להפנות את החולים גם לרופא גנטיקאי. |
| | | | |
| − | חולים עם הופעה קלאסית מציגים את הסימנים הבאים המערבים מספר מערכות ואילו חולים קלים יותר יציגו רק חלק מהסימנים: | + | סימני המחלה מתחילים להופיע בגיל הילדות ומחמירים עם הזמן גם בגברים וגם בנשים (8). סימנים ראשונים מופיעים בבנים כבר בגילאי 10-3 ובבנות בגילאי 19-13. האבחנה הסופית היא בממוצע גילאי 26 בגברים ו-32-30 בנשים (11). בתקופה זו ולמעשה עוד בשלבים העובריים מצטבר GL3 ברקמות ונוצר נזק שעלול להיות בלתי הפיך בגיל מבוגר יותר. חולים עם פעילות אנזימתית ירודה או לא קיימת מציגים תמונה קלאסית של המחלה ואילו חולים עם פעילות אנזימתית שאריתית מציגים מחלה קלה יותר או Variant diseaseנ(14). |
| | | | |
| − | ===ממצאים הקשורים למערכת העצבים===
| + | חולים עם הופעה קלאסית מציגים את הסימנים הבאים, המערבים מספר מערכות, ואילו חולים קלים יותר יציגו רק כמה מהסימנים: |
| | | | |
| − | *כאבים כרוניים (Acroparesthesia) או התקפיים המכונים "משברי פברי" ומופיעים במיוחד בזמן מאמץ, בזמן מחלת חום או במזג אוויר חם
| + | ממצאים נוירולוגיים: כאבים כרוניים (Acroparesthesia) או התקפי כאבים ("משברי פברי") במיוחד בזמן מאמץ, בזמן מחלת חום או במזג אוויר חם, שינויים בחומר הלבן של המוח, אשר ניתנים לאבחון רק ב-MRI. חולים יכולים להופיע עם תמונה קלינית של שבץ מוח או TIA (Transient Ischemic attack) בגיל צעיר יחסית (7). נמצא כי 5% מהגברים הסובלים מ-acute cryptogenic stroke הם חולי פברי שלא אובחנו (9). |
| − | *שינויים בחומר הלבן של המוח, אשר ניתנים לאבחון רק בתהודה מגנטית (MRI)
| |
| − | *חולים יכולים להופיע עם תמונה קלינית של [[שבץ מוחי]] או [[התקף איסכמי חולף]] (([[TIA]]) {{כ}} Transient Ischemic attack) בגיל צעיר יחסית. נמצא כי 5% מהגברים הסובלים מ-Acute cryptogenic stroke הם חולי אנדרסון-פברי שלא אובחנו (13).
| |
| | | | |
| − | ===ממצאים כליתיים===
| + | ממצאים כלייתיים: סימנים ראשונים הם פרוטאינוריה (proteinuria) ואיזוטנוריה (isosthenuria). עם הזמן הפגיעה מתקדמת עם עלייה בקראטינין וירידה בקצב הסינון הגלומרולרי (glomerular filtration rate - GFR). במחקרים אשר בהם נסרקו אוכלוסיות המטופלות בהמודיאליזה לנשאות מחלת פברי, נמצא כי 1%-0.25% מהגברים המטופלים בהמודיאליזה חולים בפברי (14). לאחרונה פורסמו הנחיות מטעם ה- European renal best practice המנחות נפרולוגים לחשוד במחלת פברי בקרב חולים עם מחלת כליה לא מוסברת (6). |
| | | | |
| − | *סימנים ראשונים הנם חלבון בשתן ([[פרוטאינוריה]]) ואיזוטנוריה (Isothenuria)
| + | ממצאים גסטרואיטסטינאלים: כאבים אחרי האוכל, שלשולים, תחושה של שובע מהיר. |
| − | *עם הזמן הפגיעה מתקדמת עם עליה בקראטינין וירידה בקצב הסינון הגלומרולרי ((Glomerular filtration rate (GFR)
| |
| − | *במחקרים אשר בהן נסרקו אוכלוסיות המטופלים בהמודיאליזה לנשאות מחלת אנדרסון-פברי, נמצא כי 0.25%-1% מהגברים המטופלים בהמודיאליזה חולים ב- Fabry{{כ}} (13,19)
| |
| | | | |
| − | ===ממצאים הקשורים למערכת העיכול===
| + | ממצאים עוריים: נגעים מסוג Angiokeratomas המופיעים במיוחד באזור החלציים ומתרבים עם הזמן, חוסר הזעה (Hypo-anhydrosis)נ(1). |
| | | | |
| − | *כאבים אחרי האוכל
| + | ממצאים קרדיווסקולריים: הפגיעה הלבבית עלולה להתבטא בצורות רבות, ביניהן הפרעות קצב (קיצור PR ללא גל δ, QRS מאורך, AV block ועוד), כאשר 20%-10% מהחולים נזקקים להשתלת קוצב קבוע. פגיעה בכלי הדם הכליליים עלולה להוביל להופעה של אנגינה פקטוריס (angina pectoris) ולאוטם שריר הלב. חולים יכולים להופיע עם תמונה של אי ספיקת לב משנית כגורם הבירור, פגיעה במסתמים או קרדיומיופטיה (cardiomyopathy). סימנים אלה יופיעו על פי רוב בגיל צעיר מן הממוצע באוכלוסיה. נמצא כי 4%-3% מהגברים הסובלים מקרדיומיופטיה הם חולי פברי שלא אובחנו (9, 12). מחלת הלב בפברי מאופיינת בהיפרטרופיה של החדר השמאלי הדומה לזו המתקבלת בחולים עם הפרטרופיה סרקומרית או לחץ דם גבוה (12). |
| − | *שלשולים
| |
| − | *תחושה של שובע מהיר
| |
| | | | |
| − | ===ממצאים עוריים===
| + | ממצאים המופיעים בעיניים: ב-90%-70% מהחולים (ללא הבדל מין) מופיעהcornea verticillata ב-corneal epithelium. אבחון ממצא זה מצריך שימוש במנורת סדק. זהו אינו ממצא סימפטומטי אך נוכחותו פטוגנומית למחלת פברי. |
| | | | |
| − | *נגעים מסוג אנגיוקרטומה (Angiokeratoma) מופיעים במיוחד באזור החלציים ומתרבים עם הזמן
| + | ממצאים נוספים: התקפי vertigo , ירידה בשמיעה, מחלת ריאות רסטרטיבית וחסימתית (1). |
| − | *מיעוט או חוסר הזעה (Hypo-anhydrosis){{כ}} (1,2,17,19).
| |
| | | | |
| − | ===ממצאים הקשורים למערכת הלב וכלי הדם===
| + | ממצאים התנהגותיים/ פסיכיאטריים: דיכאון שכיח מאוד בחולי פברי ומופיע בכ-46% מהחולים. כמו כן נראה ירידה בתפקוד במשימות ניהוליות, ביכולת הקשב ובמהירות עיבוד האינפורמציה תוך שמירה על הזיכרון והאינטלקט, התפישה ויכולת השיום (15). |
| | | | |
| − | ;הפגיעה הלבבית עשויה להתבטא בצורות רבות ביניהן:
| + | [[קובץ:פבריתרשים1.JPG|מרכז|ממוזער|400px|תרשים מספר 1]] |
| − | *הפרעות קצב (קיצור PR ללא גל δ, QRS מאורך, AV block ועוד), כאשר 10%-20% מהחולים נזקקים להשתלת קוצב קבוע
| |
| − | *פגיעה בכלי הדם הכליליים עשויה להוביל להופעה של [[אנגינה פקטוריס] (Angina pectoris) ול[[אוטם שריר הלב]]
| |
| − | *חולים יכולים להופיע עם תמונה של [[אי ספיקת לב]] משנית כגורם הבירור, פגיעה במסתמים או [[קרדיומיופתיה]] (Cardiomyopathy). סימנים אלה יופיעו על פי רוב בגיל צעיר מן הממוצע באוכלוסיה. נמצא כי 3%-4% מהגברים הסובלים מקרדיומיופתיה הם חולי אנדרסון-פברי שלא אובחנו (13,17). מחלת הלב באנדרסון-פברי מאופיינת בהיפרטרופיה של החדר השמאלי הדומה לזו המתקבלת בחולים עם היפרטרופיה סרקומרית או לחץ דם גבוה (18).
| |
| | | | |
| − | ===ממצאים עיניים===
| + | [[קובץ:פבריתרשים2.JPG|מרכז|ממוזער|400px|תרשים מספר 2]] |
| − | | |
| − | *ל-70%-90% מהחולים (ללא הבדל מין) מופיעה Cornea verticillata ב-Corneal epithelium. אבחון ממצא זה מצריך שימוש במנורת סדק. זהו אינו ממצא סימפטומטי אך נוכחותו פתוגנומית לאנדרסון-פברי.
| |
| − | | |
| − | ===ממצאים נוספים===
| |
| − | | |
| − | *התקפי סחרור ([[ורטיגו]])
| |
| − | *[[ירידה בשמיעה]]
| |
| − | *מחלת ריאות רסטריקטיבית וחסימתית (1,2,17,18).
| |
| | | | |
| | [[קובץ:פבריתמונה1.JPG|מרכז|ממוזער|400px|תמונה מס' 1: סימני מחלת פברי]] | | [[קובץ:פבריתמונה1.JPG|מרכז|ממוזער|400px|תמונה מס' 1: סימני מחלת פברי]] |
| | | | |
| − | ===נשאיות יכולות להיות סימפטומטיות=== | + | ==ביבליוגרפיה== |
| − | | |
| − | תוחלת החיים של נשים נשאיות למחלת אנדרסון-פברי קצרה ב- 15 שנים יחסית לממוצע באוכלוסיה, ועל פי רוב הן מפתחות סימפטומים של המחלה עם הזמן (8). הופעת הסימפטומים בנשים מופיעה בין 5 ל-10 שנים מאוחר יותר לעומת גברים, והאבחנה מתאחרת בהתאם (8,9,16). נמצא כי ב-20%-30% מהנשים מופיעים סימפטומים קשים כגון: כאבים נוירופתיים, מחלת כליות, אירועים מוחיים ומחלת לב (9). חשוב אם כן לזכור כי יש לעקוב גם אחר נשים א-סימפטומטיות שכן לא ניתן לחזות מתי יתפתחו הסימפטומים.
| |
| − | | |
| − | ==אבחנה==
| |
| − | | |
| − | ===אישור האבחנה במעבדה===
| |
| | | | |
| − | אישור האבחנה בגברים מבוסס על בדיקת פעילות האנזים אלפא גלקטוזידאז בלויקוציטים, בסרום או בפלזמה. בנשים רמת הפעילות האנזימטית יכולה להיות נמוכה או תקינה בהתאם לכמות הלויקוציטים בהם חלה אינאקטיבציה של כרומוזום ה-X{{כ}} (10). לפיכך מבוצע אישור האבחנה הקלינית בנשים על ידי בדיקת רצף הגן המקודד לאלפא גלקטוזידאז. במידה ואין ידע מוקדם על המוטציה הקיימת במשפחה מבוצע ריצוף של כל הגן. חשוב לבדוק גם נוכחות של חסר בגן על ידי בדיקת MLPA {{כ}} (Multiplex ligation-dependent probe amplification).
| + | <blockquote> |
| | + | <div style="text-align: left; direction: ltr"> |
| | | | |
| − | ===אבחון טרום לידתי===
| + | {{הערות שוליים}} |
| − | | |
| − | מומלץ להפנות את החולים והמשפחות לייעוץ גנטי כדי שיקבלו מידע על אפשרויות לאבחון טרום לידתי. אבחון זה יתבצע על ידי בדיקת הפעילות האנזימתית בעוברים זכרים או בדיקת המוטציה המשפחתית.
| |
| − | | |
| − | את הבדיקה ניתן לבצע ב[[סיסי השיליה]] בשבוע ה-11 או בתאי [[מי שפיר]] בשבוע ה-17. במידה והמוטציה ידועה ניתן גם להציע למשפחות אבחון טרם השרשה (([[PGD]]) {{כ}} Preimplantation genetic diagnosis) על ידי בדיקת תא של עוברים מ[[הפריה חוץ גופית]] ((IVF) {{כ}} In vitro fertilization).
| |
| − | | |
| − | ==טיפול==
| |
| − | | |
| − | בטרם פותח הטיפול באנזים חליפי, היה הטיפול בחולי פברי טיפול תומך וטיפול סימפטומטי בלבד ותוחלת החיים הממוצעת בגברים היתה 41 שנים (2). משנת 2001 זמינים שני תכשירים שפותחו בשיטות של הנדסה גנטית: [[Replagal]] {{כ}} ([[Agalsidase alpha]]) (רפלגל; אגלזידז אלפא) המיוצר בתאים פיברובלסטים ממקור אנושי ו-[[Fabrazyme]] {{כ}} ([[Agalsidase beta]]) (פברזיים ; אגלזידז בטא) המיוצר בתאי שחלה של אוגר סיני((CHO cells) {{כ}} Chinese hamster ovary cells).
| |
| − | | |
| − | בשנת 2001 אושר שימוש בשני התכשירים לשיווק באירופה ואילו בארה"ב אישרה רשות המזון והתרופות (FDA) ב-2003 את [[t:פברזיים - Fabrazyme|פברזיים]] בלבד.
| |
| − | | |
| − | בישראל מאושרות שתי התרופות ושתיהן כלולות בסל הבריאות לטיפול בחולי אנדרסון-פברי. [[t:רפלגל - Replagal|רפלגל]] מומלץ לשימוש במינון של 0.2mg/kg ואילו [[t:פברזיים - Fabrazyme|פברזיים]] מומלץ לשימוש במינון של 1mg/kg. שניהם ניתנים בעירוי לווריד בתדירות של פעם בשבועיים (3,5), ומחירם דומה. בשלב ראשון החולים מטופלים במסגרת בית חולים ובהמשך ובהעדר תגובות רגישות ניתן לעבור לטיפול במסגרת טיפולי בית, לאחר קבלת אישור הרופא המטפל.
| |
| − | | |
| − | בבדיקות ביוכימיות שבוצעו נראה כי רצף חומצות האמינו זהה בשני התכשירים וכך גם הפעילות האנזימתית. קיים וויכוח בספרות לגבי היתרונות של תרופה אחת על פני השנייה (20,21,22).
| |
| − | | |
| − | מאחר ומדובר בהזרקה של חלבון אשר בחולים מסוימים הנו זר לגוף, תתכן יצירת נוגדנים כנגד האנזים המוזרק (23,24). דווח כי ב-30% מבין החולים המתפתחים נוגדנים מסוג IgG נגד האנזים ונשארים הנוגדנים לאורך זמן, אולם השפעתם על יעילות הטיפול עדין לא ברורה (20,21). נשים הטרוזיגוטיות אינן מפתחות נוגדנים ותופעות לוואי בדרך כלל.
| |
| − | | |
| − | ===במי לטפל ומתי להתחיל טיפול===
| |
| − | | |
| − | ההמלצה לטיפול בחולים זכרים הנה להתחיל בטיפול מוקדם ככל האפשר על מנת למנוע את נזקי האגירה בתאים וברקמות. מן הראיות שהצטברו עד כה נראה כי חשוב לעקוב אחר התפתחות המחלה בנשים "נשאיות של המוטציה" ולשקול טיפול בשלבים מוקדמים יחסית עם הופעת הסימפטומים כדי למנוע נזק בלתי הפיך (1,4). בילדים יש וויכוח אם להתחיל טיפול לפני הופעת התסמינים או להמתין להופעת הסימנים הקליניים (15).
| |
| − | | |
| − | הטיפול כולל מעקב על ידי צוות רב תחומי, הכולל את רופא המשפחה, נפרולוג, קרדיולוג, נוירולוג, מומחה לכאבים, ומומחים נוספים בהתאם לצורך. יש לשים דגש מיוחד על מעקב התפקוד הכילייתי, התפקוד הלבבי ומערכת העצבים. מומלץ טיפול סימפטומטי תומך כמו טיפול במעכבי אנזים המהפך (Angiotensin converting enzyme (ACE) inhibitors) ובחוסמי הקולטן לאנגיוטנסין ((ARBs) {{כ}} Angiotensin Receptor Blockers) בחולים עם [[פרוטאינוריה]], [[יתר לחץ דם]] ו[[הפרעות בשומני הדם]] (דיסליפידמיה). ניתן לטפל ב-[[Aspirin]] ([[אספירין]]) למניעת [[קרישיות יתר]].
| |
| − | | |
| − | בגלל אופייה הפרוגרסיבי של המחלה, חשוב לזכור, כי המפתח לטיפול יעיל והטבת איכות חיי החולים נעוץ באבחון המוקדם של המחלה ובתחילת מתן האנזים החליפי בטרם נוצר נזק בלתי הפיך לרקמה.
| |
| − | | |
| − | ==ביבליוגרפיה==
| |
| | | | |
| − | <div align=left style="margin-right: 50px;">
| |
| − | # Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Grabowski G, Packman S, Wilcox WR. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338-46.
| |
| − | # Breunig F, Wanner C. J Nephrol. Update on Fabry disease: kidney involvement, renal progression and enzyme replacement therapy. 2008; 21(1):32-7.
| |
| − | # Lidove O, Joly D, Barbey F, Bekri S, Alexandra JF, Peigne V, Jaussaud R, Papo T. Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature. Int J Clin Pract. 2007; 61(2):293-302.
| |
| − | # Ortiz A, Oliveira JP, Wanner C, Brenner BM, Waldek S, Warnock DG. Recommendations and guidelines for the diagnosis and treatment of Fabry nephropathy in adults. Nat Clin Pract Nephrol. 2008; 4(6):327-36. Epub 2008 Apr 22.
| |
| − | # Desnick RJ. Enzyme replacement therapy for Fabry disease: lessons from two alpha-galactosidase-A A orphan products and one FDA approval .Expert Opin Biol Ther. 2004; 4(7):1167-76.
| |
| − | # Stratta P, Quaglia M, Messina M, Cavagnino A, Ragazzoni E, Bergamo D, Mazzucco G. The challenges of diagnosing Fabry disease. Am J Kidney Dis. 2008; 51(5):860-4.
| |
| − | # https://research.cchmc.org/LOVD/home.php?select_db=GLA
| |
| − | # Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. 2007; 9(1):34-45.
| |
| − | # Hughes DA. Early therapeutic intervention in females with Fabry disease. Acta Paediatr Suppl. 2008; 97(457):41-7.
| |
| − | # Dobyns WB. The pattern of inheritance of X-linked traits is not dominant or recessive, just X-linked. Acta Paediatr Suppl. 2006; 95(451):11-5.
| |
| − | # Maier EM, Osterrieder S, Whybra C, Ries M, Gal A, Beck M, Roscher AA, Muntau AC. Disease manifestations and X inactivation in heterozygous females with Fabry disease. Acta Paediatr Suppl. 2006; 95 (451):30-8.
| |
| − | # Ropers HH, Wienker TF, Grimm T, Schroetter K, Bender K. Evidence for preferential X-chromosome inactivation in a family with Fabry disease. Am J Hum Genet. 1977; 29 (4):361-70.
| |
| − | # Marco Spada, Severo Pagliardini, Makiko Yasuda, Turgut Tukel, Geetha Thiagarajan, Hitoshi Sakuraba, Alberto Ponzone, and Robert J. Desnick. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening. Am. J. Hum. Genet. 2006; 79:31–40.
| |
| − | # Hwu WL, Chien YH, Lee NC, Chiang SC, Dobrovolny R, Huang AC, Yeh HY, Chao MC, Lin SJ, Kitagawa T, Desnick RJ, Hsu LW. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum Mutat. 2009;30(10):1397-405.
| |
| − | # Hopkin RJ, Bissler J, Banikazemi M, Clarke L, Eng CM, Germain DP, Lemay R, Tylki-Szymanska A, Wilcox WR. Characterization of Fabry Disease in 352 Pediatric Patients in the Fabry Registry. Pediatr Res. 2008.
| |
| − | # Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, Sims K, Waldek S, Pastores GM, Lee P, Eng CM, Marodi L, Stanford KE, Breunig F, Wanner C, Warnock DG, Lemay RM, Germain DP. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008; 93 (2):112-28.
| |
| − | # Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ, Elliott PM. Prevalence of Anderson- Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002; 105:1407–11.
| |
| − | # Gambarin FI, Disabella E, Narula J, Diegoli M, Grasso M, Serio A, Favalli BM, Agozzino M, Tavazzi L, Fraser AG, Arbustini E. When should cardiologists suspect Anderson-Fabry disease?. Am J Cardiol. 2010 15;106(10):1492-9.
| |
| − | # Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, Kanzaki T, Enriquez AL, Eng CM, Tanaka H, Tei C, Desnick RJ. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a "renal variant" phenotype. Kidney Int. 2003; 64:801–7.
| |
| − | # Vedder AC, Breunig F, Donker-Koopman WE, Mills K, Young E, Winchester B, Ten Berge IJ, Groener JE, Aerts JM, Wanner C, Hollak CE.; Treatment of Fabry disease with different dosing regimens of agalsidase: effects on antibody formation and GL-3. Mol Genet Metab. 2008 Jul; 94(3):319-25.
| |
| − | # Vedder AC, Linthorst GE, Houge G, Groener JE, Ormel EE, Bouma BJ, Aerts JM, Hirth A, Hollak CE. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS ONE. 2007; 2(7): 598.
| |
| − | # Mehta A, Beck M, Kampmann C, Frustaci A, Germain DP, Pastores GM, Sunder-Plassmann G. Enzyme replacement therapy in Fabry disease: Comparison of agalsidase alfa and agalsidase beta. Mol Genet Metab. 2008
| |
| − | # Bodensteiner D, Scott CR, Sims KB, Shepherd GM, Cintron RD, Germain DP. uccessful reinstitution of [[t:Agalsidase beta|Agalsidase beta]] therapy in Fabry disease patients with previous IgE-antibody or skin-test reactivity to the recombinant enzyme.Genet Med. 2008;10(5):353-8.
| |
| − | # Ohashi T, Iizuka S, Ida H, Eto Y. Reduced alpha-Gal A enzyme activity in Fabry fibroblast cells and Fabry mice tissues induced by serum from antibody positive patients with Fabry disease. Mol Genet Metab. 2008; 94(3):313-8.
| |
| | </div> | | </div> |
| | + | </blockquote> |
| | | | |
| | ==קישורים חיצוניים== | | ==קישורים חיצוניים== |
| מחלת אנדרסון-פברי
|
| Anderson-Fabry disease
|
|
Alpha galactosidase - the protein that is deficient in Fabry disease
|
| שמות נוספים
|
Alpha-galactosidase A deficiency, Angiokeratoma corporis diffusum, Ceramide trihexosidosis, Ruiter-Pompen-Wyers syndrome, Sweeley-Klionsky disease
|
|
| ICD-10
|
Chapter E 75.2
|
|
| ICD-9
|
272.7
|
|
| MeSH
|
D000795
|
|
| יוצר הערך
|
פרופסור אניק רז-רוטשילד
|
|
|
|
לערכים נוספים הקשורים לנושא זה, ראו את דף הפירושים – מחלות אגירה
מחלת אנדרסון-פברי (Anderson-Fabry) היא מחלת אגירה ליזוזומלית המורשת בתאחיזה לכרומוזום ה-X (X- linked). תפוצתה באוכלוסיה היא פן- אתנית ושכיחותה אינה ברורה.
מחלת פברי מאופיינת על ידי ירידה או חסר בפעילות של האנזים הליזוזומלי alpha-galactosidase-A המובילה לאגירה של סובסטרט ממשפחת הגליקוספינגוליפידים ולפגיעה רב מערכית. מחלת Fabry היא חלק מקבוצת הסיפינוליפידוזות .(sphingolipidosis) הפגיעות העיקריות הן במערכת העצבים, במערכת הקרדיווסקולרית ובכליות. הגברים הינם המיזיגוטים (hemizygotes) ומפתחים מחלה רב מערכתית ואילו הנשים הן הטרוזיגוטיות אך על פי רוב תפתחנה גם הן תסמינים אשר עלולים להיות חמורים כמו בגברים.
זוהי מחלה פרוגרסיבית וסימניה הראשונים יכולים להופיע כבר בגיל הילדות. חומרת המחלה וצורת הופעתה מגוונות בין המשפחות השונות וגם בתוך כל משפחה.
זה כבר יותר מ-14 שנה קיים טיפול באנזים חליפי ועל כן, אבחון מוקדם ומתן טיפול, בטרם נוצר נזק בלתי הפיך לרקמה ולאיברים, עשויים לשפר את איכות חיי החולים ואולי אף להאריכם [1].
שמות נוספים: Fabry disease ,Fabry’s disease ,Alpha-galactosidase A deficiency, Angiokeratoma corporis diffusum, Ceramide trihexosidosis, Ruiter-Pompen-Wyers syndrome, Sweeley-Klionsky disease.
פתופיזיולוגיה
על פי הידוע נכון להיום, בעקבות חסר או ירידה בפעילות האנזים alpha-galactosidase-A נמנע פירוק תקין של גליקוספינגוליפידים (Glycosphingolipids) ובעיקר Globotriaosylceramide (GL3,Gb3, ceramide trihexoside) בתאים שונים ובמיוחד בולט הדבר בתאי האנדותל הווסקולרי. הסובסטרט נאגר בליזוזומים וגורם לפגיעות משניות בתפקוד תאים כגון תאי עצב, תאי האפיתל הגלומרולרי (glomerular epithelia) והטובולרי (tubular epithelia) של הכליות, תאי מערכת ההולכה של קוצבי הלב וקרדיומיוציטים (cardiomyocytes)נ(1). הפגיעה התאית גוררת פגיעה ברקמה עם הופעה של איסכמיה (ischemia) ופיברוזיס (fibrosis). עד היום לא ברור מתי הנזק לתאים הוא בלתי הפיך ומתי הטיפול אינו יכול להביא לשיפור במצבם של החולים. האגירה מתחילה כבר בשלב העוברי [2] ומצטברות עדויות כי מתן טיפול מוקדם הוא יעיל [1], {{הערה|שם=הערה5|}, [3].
תורשה ושכיחות
הגן המקודד לאנזים alpha-galactosidase-A ממופה על גבי הזרוע הארוכה של כרומוזום X [Xq22] ומכיל 7 אקסונים [4]. לכל משפחה מוטציה משלה (private mutation). מחלת פברי מורשת בתאחיזה לכרומוזום ה-X. בניגוד למחלות תורשתיות אחרות המועברות בתאחיזה לכרומוזום ה-X, שבהן הנשים הנשאיות בדרך כלל אינן סימפטומטיות, נשים הנושאות מוטציות בגן ל-Alpha galactosidase-A יהיו עם הזמן סימפטומטיות בדרגות חומרה שונות [1]. כמו כן, בשל דפוס ההורשה האחוז ל-X, תהיינה בנותיו של גבר חולה פברי בהכרח נשאיות ואילו כל בניו יהיו בריאים. לגבי אישה נשאית, לכל בת יהיה סיכוי של 50% להיות נשאית עם אפשרות לפתח את המחלה, כאשר לכל בן של אישה נשאית, הסיכוי לבן חולה עומד על 50%.
מחלת Fabry אינה שכיחה יותר בעדות או בעמים מסוימים ופוגעת בכל האוכלוסיה בשכיחות דומה. בעבר תוארה המחלה כמחלה נדירה המופיעה בזכרים בשכיחות של 1:40,000-60,000 [1], אולם במחקרי סקר ילודים שנערכו במספר מדינות, נמצאה שכיחות גבוהה בהרבה 1:1237-1:13,341 [5]. בישראל מאובחנים כיום כ-30-25 חולים (גברים, נשים וילדים).
סימני המחלה
רוב התסמינים שמהם סובלים חולי פברי אינם ספציפיים למחלה ולכן אבחון חולה עם פברי ואבחון חולים עם מחלות נדירות בכלל מהווים אתגר משמעותי.
חולה פברי יופיע במרפאה עם אחד או יותר מהממצאים הבאים (תמונה מס' 1): נוכחות נגעים עוריים מסוג angiokeratoma, כאבים "שורפים" בגפיים (acroparesthesia), כאבי בטן, שלשולים במיוחד אחרי אכילה, חוסר הזעה, עייפות, אי סבילות לחום, אי סבילות למאמץ וכן סיפור משפחתי של תסמינים כמו אי ספיקת כליות, שבץ מוחי (stroke), או מחלה קרדיאלית (1).
אחד הכלים היעילים ביותר לאבחון המחלה הוא בירור האם קיימים קרובי משפחה עם תסמינים אשר יכולים להיות קשורים למחלת פברי. כלומר, בניית עץ משפחה המתאים לתורשה בתאחיזה ל-X שבו נמצא חולים עם התסמינים המוזכרים שהקשר ביניהם הוא הנשים במשפחה. לאחר אבחון של אדם במשפחה חדשה, יש צורך לבדוק את כל בני המשפחה המצויים בסיכון על בסיס נתוני המשפחה, תוך התייחסות לאופן ההורשה. לשם כך מומלץ להפנות את החולים גם לרופא גנטיקאי.
סימני המחלה מתחילים להופיע בגיל הילדות ומחמירים עם הזמן גם בגברים וגם בנשים (8). סימנים ראשונים מופיעים בבנים כבר בגילאי 10-3 ובבנות בגילאי 19-13. האבחנה הסופית היא בממוצע גילאי 26 בגברים ו-32-30 בנשים (11). בתקופה זו ולמעשה עוד בשלבים העובריים מצטבר GL3 ברקמות ונוצר נזק שעלול להיות בלתי הפיך בגיל מבוגר יותר. חולים עם פעילות אנזימתית ירודה או לא קיימת מציגים תמונה קלאסית של המחלה ואילו חולים עם פעילות אנזימתית שאריתית מציגים מחלה קלה יותר או Variant diseaseנ(14).
חולים עם הופעה קלאסית מציגים את הסימנים הבאים, המערבים מספר מערכות, ואילו חולים קלים יותר יציגו רק כמה מהסימנים:
ממצאים נוירולוגיים: כאבים כרוניים (Acroparesthesia) או התקפי כאבים ("משברי פברי") במיוחד בזמן מאמץ, בזמן מחלת חום או במזג אוויר חם, שינויים בחומר הלבן של המוח, אשר ניתנים לאבחון רק ב-MRI. חולים יכולים להופיע עם תמונה קלינית של שבץ מוח או TIA (Transient Ischemic attack) בגיל צעיר יחסית (7). נמצא כי 5% מהגברים הסובלים מ-acute cryptogenic stroke הם חולי פברי שלא אובחנו (9).
ממצאים כלייתיים: סימנים ראשונים הם פרוטאינוריה (proteinuria) ואיזוטנוריה (isosthenuria). עם הזמן הפגיעה מתקדמת עם עלייה בקראטינין וירידה בקצב הסינון הגלומרולרי (glomerular filtration rate - GFR). במחקרים אשר בהם נסרקו אוכלוסיות המטופלות בהמודיאליזה לנשאות מחלת פברי, נמצא כי 1%-0.25% מהגברים המטופלים בהמודיאליזה חולים בפברי (14). לאחרונה פורסמו הנחיות מטעם ה- European renal best practice המנחות נפרולוגים לחשוד במחלת פברי בקרב חולים עם מחלת כליה לא מוסברת (6).
ממצאים גסטרואיטסטינאלים: כאבים אחרי האוכל, שלשולים, תחושה של שובע מהיר.
ממצאים עוריים: נגעים מסוג Angiokeratomas המופיעים במיוחד באזור החלציים ומתרבים עם הזמן, חוסר הזעה (Hypo-anhydrosis)נ(1).
ממצאים קרדיווסקולריים: הפגיעה הלבבית עלולה להתבטא בצורות רבות, ביניהן הפרעות קצב (קיצור PR ללא גל δ, QRS מאורך, AV block ועוד), כאשר 20%-10% מהחולים נזקקים להשתלת קוצב קבוע. פגיעה בכלי הדם הכליליים עלולה להוביל להופעה של אנגינה פקטוריס (angina pectoris) ולאוטם שריר הלב. חולים יכולים להופיע עם תמונה של אי ספיקת לב משנית כגורם הבירור, פגיעה במסתמים או קרדיומיופטיה (cardiomyopathy). סימנים אלה יופיעו על פי רוב בגיל צעיר מן הממוצע באוכלוסיה. נמצא כי 4%-3% מהגברים הסובלים מקרדיומיופטיה הם חולי פברי שלא אובחנו (9, 12). מחלת הלב בפברי מאופיינת בהיפרטרופיה של החדר השמאלי הדומה לזו המתקבלת בחולים עם הפרטרופיה סרקומרית או לחץ דם גבוה (12).
ממצאים המופיעים בעיניים: ב-90%-70% מהחולים (ללא הבדל מין) מופיעהcornea verticillata ב-corneal epithelium. אבחון ממצא זה מצריך שימוש במנורת סדק. זהו אינו ממצא סימפטומטי אך נוכחותו פטוגנומית למחלת פברי.
ממצאים נוספים: התקפי vertigo , ירידה בשמיעה, מחלת ריאות רסטרטיבית וחסימתית (1).
ממצאים התנהגותיים/ פסיכיאטריים: דיכאון שכיח מאוד בחולי פברי ומופיע בכ-46% מהחולים. כמו כן נראה ירידה בתפקוד במשימות ניהוליות, ביכולת הקשב ובמהירות עיבוד האינפורמציה תוך שמירה על הזיכרון והאינטלקט, התפישה ויכולת השיום (15).
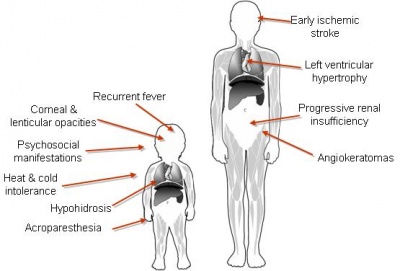
תמונה מס' 1: סימני מחלת פברי
ביבליוגרפיה
- ↑ 1.0 1.1 1.2 1.3 שגיאת ציטוט: תג
<ref> לא תקין; לא נכתב טקסט עבור הערות השוליים בשם הערה1
- ↑ שגיאת ציטוט: תג
<ref> לא תקין; לא נכתב טקסט עבור הערות השוליים בשם הערה2
- ↑ שגיאת ציטוט: תג
<ref> לא תקין; לא נכתב טקסט עבור הערות השוליים בשם הערה6
- ↑ שגיאת ציטוט: תג
<ref> לא תקין; לא נכתב טקסט עבור הערות השוליים בשם הערה5
- ↑ שגיאת ציטוט: תג
<ref> לא תקין; לא נכתב טקסט עבור הערות השוליים בשם הערה8
קישורים חיצוניים
פורסם בגיליון מס' 2 בנושא מחלות יתומות, פברואר 2016, medic-digital
המידע שבדף זה נכתב על ידי פרופ' אניק רז-רוטשילד - מנהלת המכון למחלות נדירות, המחלקה לגנטיקה, המרכז הרפואי שיבא, תל השומר

 כניסה
כניסה  עקבו אחרינו בפייסבוק
עקבו אחרינו בפייסבוק 


